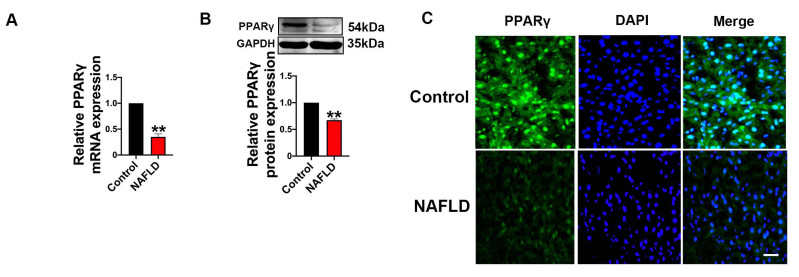
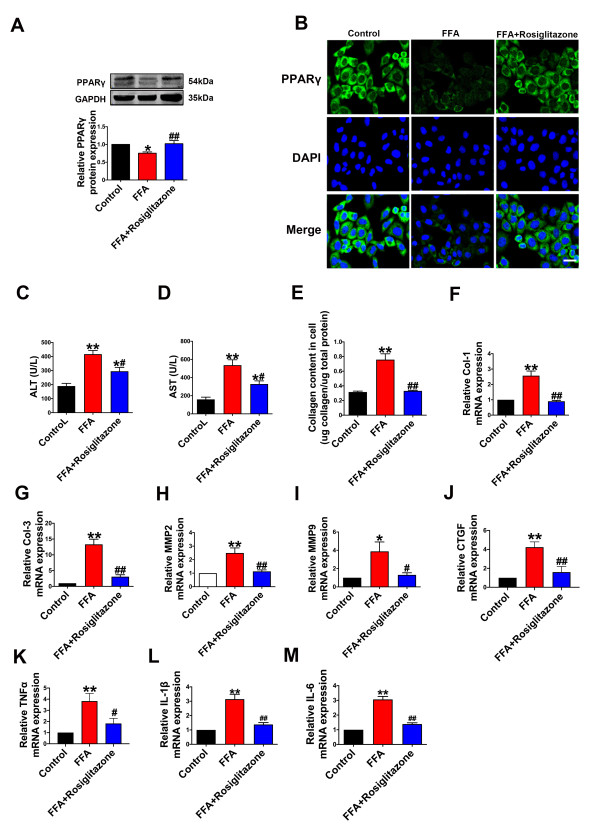
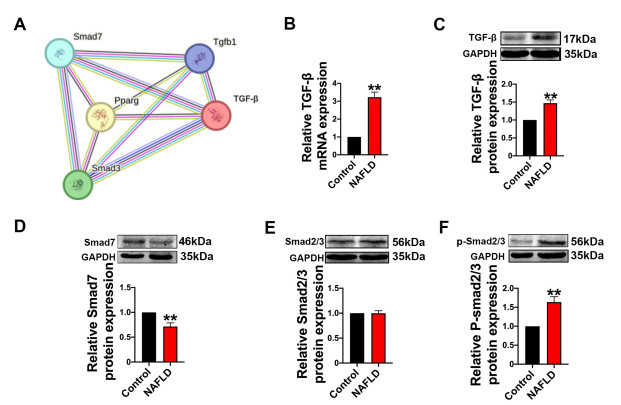
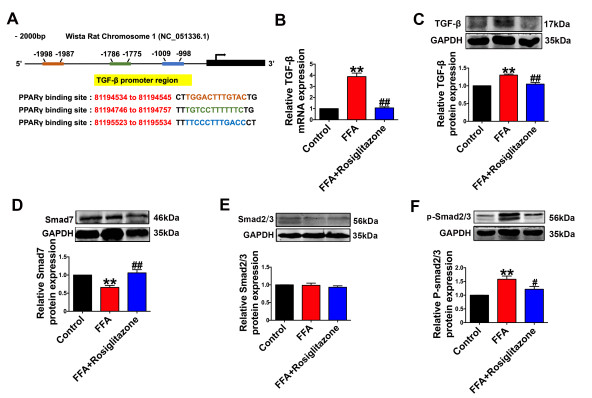
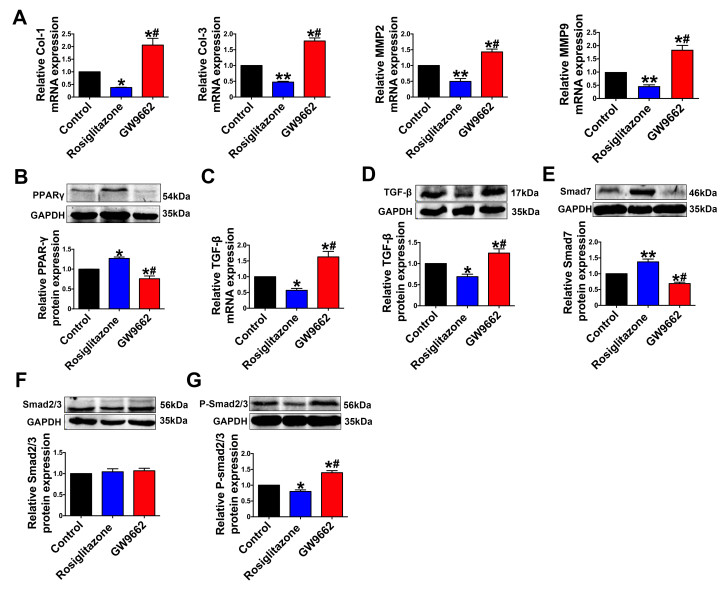
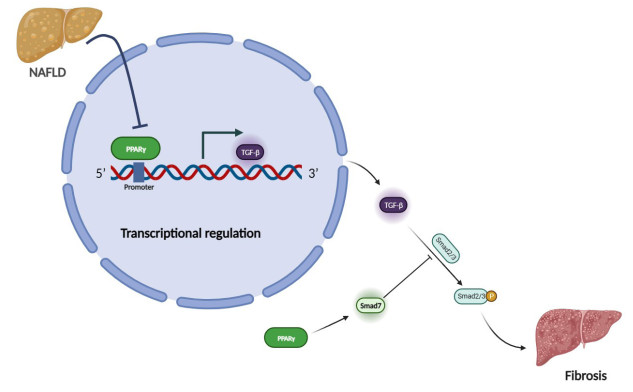
| Citation: | Qingwei Zhang, Wenjie Zhao, Zeqi Sun, Xinxin Dong, Liwei Zhu, Zhen Zhang, Ximing Chen, Yingying Hu, Menghan Du, Jiamin Li, Yong Zhang. Peroxisome proliferator-activated receptors gama ameliorates liver fibrosis in non-alcoholic fatty liver disease by inhibiting TGF-β/Smad signaling activation[J]. Frigid Zone Medicine, 2024, 4(1): 12-22. doi: 10.2478/fzm-2024-0002

|

| [1] |
Le M H, Le D M, Baez T C, et al. Global incidence of non-alcoholic fatty liver disease: A systematic review and meta-analysis of 63 studies and 1, 201, 807 persons. J Hepatol, 2023; 79(2): 287-295. doi: 10.1016/j.jhep.2023.03.040
|
| [2] |
Chen H, Zhan Y, Zhang J, et al. The global, regional, and national burden and trends of NAFLD in 204 Countries and territories: an analysis from global burden of disease 2019. JMIR Public Health Surveill, 2022; 8(12): e34809. doi: 10.2196/34809
|
| [3] |
Xia M, Sun X, Zheng L, et al. Regional difference in the susceptibility of non-alcoholic fatty liver disease in China. BMJ Open Diabetes Res Care, 2020; 8(1): e001311. doi: 10.1136/bmjdrc-2020-001311
|
| [4] |
Liu S, Liu Y, Wan B, et al. Association between vitamin D status and non-alcoholic fatty liver disease: a population-based study. J Nutr Sci Vitaminol (Tokyo), 2019; 65: 303-308. doi: 10.3177/jnsv.65.303
|
| [5] |
Jain R, Wade G, Ong I, et al. Determination of tissue contributions to the circulating lipid pool in cold exposure via systematic assessment of lipid profiles. J Lipid Res, 2022; 63(7): 100197. doi: 10.1016/j.jlr.2022.100197
|
| [6] |
Su D, Zhou T, Wang Y, et al. Cold exposure regulates hepatic glycogen and lipid metabolism in newborn goats. Int J Mol Sci, 2023; 24(18): 14330. doi: 10.3390/ijms241814330
|
| [7] |
Heda R, Yazawa M, Shi M, et al. Non-alcoholic fatty liver and chronic kidney disease: Retrospect, introspect, and prospect. World J Gastroenterol, 2021; 27(17): 1864-1882. doi: 10.3748/wjg.v27.i17.1864
|
| [8] |
Wang S, Tang C, Zhao H, et al. Network pharmacological analysis and experimental validation of the mechanisms of action of Si-Ni-San against liver fibrosis. Front Pharmacol, 2021; 12: 656115. doi: 10.3389/fphar.2021.656115
|
| [9] |
Wei M, Yan X, Xin X, et al. Hepatocyte-specific Smad4 deficiency alleviates liver fibrosis via the p38/p65 pathway. Int J Mol Sci, 2022; 23(19): 11696. doi: 10.3390/ijms231911696
|
| [10] |
Li Z, Wang Z, Dong F, et al. Germacrone attenuates hepatic stellate cells activation and liver fibrosis via regulating multiple signaling pathways. Front Pharmacol, 2021; 12: 745561. doi: 10.3389/fphar.2021.745561
|
| [11] |
Yuan S, Wei C, Liu G, et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1alpha/SLC7A11 pathway. Cell Prolif, 2022; 55(1): e13158. doi: 10.1111/cpr.13158
|
| [12] |
Seo H Y, Lee S H, Han E, et al. Evogliptin directly inhibits inflammatory and fibrotic signaling in isolated liver cells. Int J Mol Sci, 2022; 23(19): 11636. doi: 10.3390/ijms231911636
|
| [13] |
Song Y, Wei J, Li R, et al. Tyrosine kinase receptor B attenuates liver fibrosis by inhibiting TGF-beta/SMAD signaling. Hepatology, 2023; 78(5): 1433-1447. doi: 10.1097/HEP.0000000000000319
|
| [14] |
Vallee A, Vallee J N, Lecarpentier Y. PPARgamma agonists: potential treatment for autism spectrum disorder by inhibiting the canonical WNT/beta-catenin pathway. Mol Psychiatry, 2019; 24(5): 643-652. doi: 10.1038/s41380-018-0131-4
|
| [15] |
Khan R S, Bril F, Cusi K, et al. Modulation of insulin resistance in nonalcoholic fatty liver disease. Hepatology, 2019; 70(2): 711-724. doi: 10.1002/hep.30429
|
| [16] |
Trauner M, Fuchs C D. Novel therapeutic targets for cholestatic and fatty liver disease. Gut, 2022; 71(1): 194-209. doi: 10.1136/gutjnl-2021-324305
|
| [17] |
Snyder H S, Sakaan S A, March K L, et al. Non-alcoholic fatty liver disease: a review of anti-diabetic pharmacologic therapies. J Clin Transl Hepatol, 2018; 6(2): 168-174. doi: 10.14218/JCTH.2017.00050
|
| [18] |
Chang E, Park C Y, Park S W. Role of thiazolidinediones, insulin sensitizers, in non-alcoholic fatty liver disease. J Diabetes Investig, 2013; 4(6): 517-524. doi: 10.1111/jdi.12107
|
| [19] |
Li J, Gong L, Zhang R, et al. Fibroblast growth factor 21 inhibited inflammation and fibrosis after myocardial infarction via EGR1. Eur J Pharmacol, 2021; 910: 174470. doi: 10.1016/j.ejphar.2021.174470
|
| [20] |
Zhang Q, Chen X, Hu Y, et al. BIRC5 inhibition is associated with pyroptotic cell death via Caspase3-GSDME pathway in lung adenocarcinoma cells. Int J Mol Sci, 2023; 24(19): 14663. doi: 10.3390/ijms241914663
|
| [21] |
Li J, Li Y, Liu Y, et al. Fibroblast growth factor 21 ameliorates Na(V)1.5 and Kir2.1 channel dysregulation in human AC16 cardiomyocytes. Front Pharmacol, 2021; 12: 715466. doi: 10.3389/fphar.2021.715466
|
| [22] |
Li J, Xu C, Liu Y, et al. Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Res Cardiol, 2020; 115(2): 9. doi: 10.1007/s00395-019-0768-4
|
| [23] |
Zhou Q Y, Yang H M, Liu J X, et al. MicroRNA-497 induced by Clonorchis sinensis enhances the TGF-beta/Smad signaling pathway to promote hepatic fibrosis by targeting Smad7. Parasit Vectors, 2021; 14(1): 472. doi: 10.1186/s13071-021-04972-3
|
| [24] |
Ganai A A, Husain M. Genistein attenuates D-GalN induced liver fibrosis/chronic liver damage in rats by blocking the TGF-beta/Smad signaling pathways. Chem Biol Interact, 2017; 261: 80-85. doi: 10.1016/j.cbi.2016.11.022
|
| [25] |
Porcuna J, Minguez-Martinez J and Ricote M. The PPARalpha and PPARgamma epigenetic landscape in cancer and immune and metabolic disorders. Int J Mol Sci, 2021; 22(19): 10573. doi: 10.3390/ijms221910573
|
| [26] |
Hendawy O, Gomaa H A M, Hussein S, et al. Cold-pressed raspberry seeds oil ameliorates high-fat diet triggered non-alcoholic fatty liver disease. Saudi Pharm J, 2021; 29(11): 1303-1313. doi: 10.1016/j.jsps.2021.09.014
|
| [27] |
Wu X, Cheng B, Guo X, et al. PPARalpha/gamma signaling pathways are involved in Chlamydia pneumoniae-induced foam cell formation via upregulation of SR-A1 and ACAT1 and downregulation of ABCA1/G1. Microb Pathog, 2021; 161(Pt B): 105284. doi: 10.1016/j.micpath.2021.105284
|
| [28] |
Shan S, Zhou J, Yin R, et al. Millet bran protein hydrolysate displays the anti-non-alcoholic fatty liver disease effect via activating peroxisome proliferator-activated receptor gamma to restrain fatty acid uptake. J Agric Food Chem, 2023; 71(3): 1628-1642. doi: 10.1021/acs.jafc.2c08169
|
| [29] |
He J, Hong B, Bian M, et al. Docosahexaenoic acid inhibits hepatic stellate cell activation to attenuate liver fibrosis in a PPARgamma-dependent manner. Int Immunopharmacol, 2019; 75: 105816. doi: 10.1016/j.intimp.2019.105816
|
| [30] |
Fiorucci S, Distrutti E. Linking liver metabolic and vascular disease via bile acid signaling. Trends Mol Med, 2022; 28(1): 51-66. doi: 10.1016/j.molmed.2021.10.005
|
| [31] |
Kim E R, Park J S, Kim J H, et al. A GLP-1/GLP-2 receptor dual agonist to treat NASH: targeting the gut-liver axis and microbiome. Hepatology, 2022; 75(6): 1523-1538. doi: 10.1002/hep.32235
|
| [32] |
Brougham-Cook A, Jain I, Kukla D A, et al. High throughput interrogation of human liver stellate cells reveals microenvironmental regulation of phenotype. Acta Biomater, 2022; 138: 240-253. doi: 10.1016/j.actbio.2021.11.015
|
| [33] |
Wu G, Liu Y, Feng W, et al. Hypoxia-induced adipose lipolysis requires fibroblast growth factor 21. Front Pharmacol, 2020; 11: 1279. doi: 10.3389/fphar.2020.01279
|
| [34] |
Chojkier M, Lyche K D, Filip M. Increased production of collagen in vivo by hepatocytes and nonparenchymal cells in rats with carbon tetrachloride-induced hepatic fibrosis. Hepatology, 1988; 8(4): 808-814. doi: 10.1002/hep.1840080419
|
| [35] |
Wehrhan F, Hyckel P, Guentsch A, et al. Bisphosphonate-associated osteonecrosis of the jaw is linked to suppressed TGFbeta1-signaling and increased Galectin-3 expression: a histological study on biopsies. J Transl Med, 2011; 9: 102. doi: 10.1186/1479-5876-9-102
|
| [36] |
Hilt Z T, Maurya P, Tesoro L, et al. Beta2M signals monocytes through non-canonical TGFbeta receptor signal transduction. Circ Res, 2021; 128(5): 655-669. doi: 10.1161/CIRCRESAHA.120.317119
|
| [37] |
Zhong C, Lin Z, Ke L, et al. Recent research progress (2015-2021) and perspectives on the pharmacological effects and mechanisms of tanshinone IIA. Front Pharmacol, 2021; 12: 778847. doi: 10.3389/fphar.2021.778847
|
| [38] |
Tian H, Liu H, Yu J, et al. PHF14 enhances DNA methylation of SMAD7 gene to promote TGF-beta-driven lung adenocarcinoma metastasis. Cell Discov, 2023; 9(1): 41. doi: 10.1038/s41421-023-00528-0
|
 fzm-4-1-12_ESM.pdf
fzm-4-1-12_ESM.pdf
|

|

Figures(7) / Tables(1)
Editorial Office: Frigid Zone Medicine, Editorial Office of Frigid Zone Medicine, Heilongjiang Health Development Research Center, Zhongshan Road 112, Xiangfang District, Harbin, 150036, China
Tel: 0451-87253028
E-mail: editorialoffice@frigidzonemedicine.com
Supported by:
Beijing Renhe Information Technology Co., Ltd.


 Submit
Submit


 DownLoad:
DownLoad: