 投稿系统
投稿系统

Computation-aided novel epitope prediction by targeting spike protein's functional dynamics in Omicron
doi: 10.2478/fzm-2023-0001
-
-

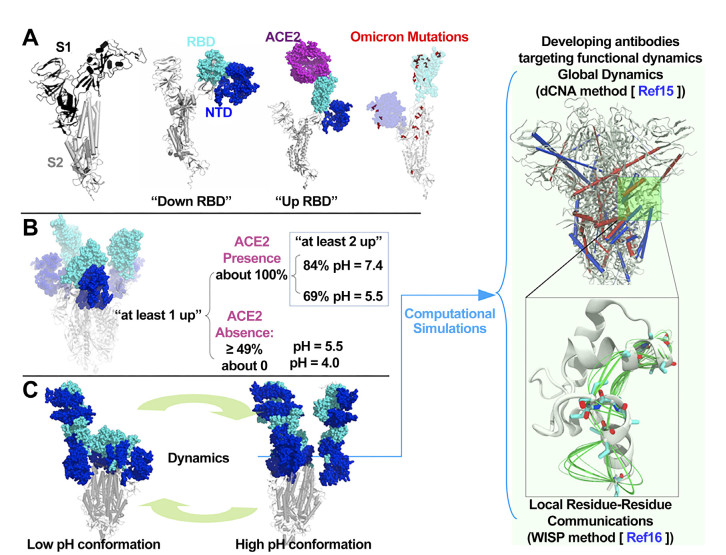
Figure 1. Sipke protein structure and dynamics
(A) The protomer of S protein showing S1 and S2 subunits. S1 contains the RBD and NTD domains. The RBD has "down" and "up" configurations, and the 'up' configuration is required for binding host ACE2; (B) Experimentally observed pH-regulated S protein conformational plasticity during the endosomal entry of SARS-CoV-2 into host cell[6]; (C) The functional dynamics underlying the pH-regulated S protein plasticity is integral to SARS-CoV-2 infectivity. Computational simulations are able to decipher such dynamics and identify conserved regions that may have antigenicity; RBD, receptor binding domain; ACE2, angiotensin conversion enzyme 2; NTD, N-terminal domain.
-
[1] Cele S, Jackson L, Khoury D S, et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature, 2022; 602(7898): 654-656. doi: 10.1038/s41586-021-04387-1 [2] Qiao J, Li Y S, Zeng R, et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science, 2021; 371(6536): 1374-1378. doi: 10.1126/science.abf1611 [3] Quan B X, Shuai H, Xia A J, et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat Microbiol, 2022; 7(5): 716–725. doi: 10.1038/s41564-022-01119-7 [4] Nemet I, Kliker L, Lustig Y, et al. Third BNT162b2 vaccination neutralization of SARS-CoV-2 Omicron infection. N Engl J Med, 2022; 386(5): 492-494. doi: 10.1056/NEJMc2119358 [5] Du X, Tang H, Gao L, et al. Omicron adopts a different strategy from Delta and other variants to adapt to host. Signal Transduct Target Ther, 2022; 7(1): 45. doi: 10.1038/s41392-022-00903-5 [6] Zhou T, Tsybovsky Y, Gorman J, et al. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-Dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe, 2020; 28(6): 867-879. e5. doi: 10.1016/j.chom.2020.11.004 [7] Triveri A, Serapian S A, Marchetti F, et al. SARS-CoV-2 spike protein mutations and escape from antibodies: a computational model of epitope loss in variants of concern. J Chem Inf Model, 2021; 61(9): 4687-4700. doi: 10.1021/acs.jcim.1c00857 [8] Weisblum Y, Schmidt F, Zhang F, et al. Escape from neutralizing antibodies 1 by SARS-CoV-2 spike protein variants. Elife, 2020; 9: e61312. doi: 10.7554/eLife.61312 [9] Wang Y, Wang L, Cao H, et al. SARS-CoV-2 S1 is superior to the RBD as a COVID-19 subunit vaccine antigen. J Med Virol, 2021;93(2): 892-898. doi: 10.1002/jmv.26320 [10] Miller N L, Clark T, Raman R, et al. Insights on the mutational landscape of the SARS-CoV-2 Omicron variant receptor-binding domain. Cell Rep Med, 2022; 3(2): 100527. doi: 10.1016/j.xcrm.2022.100527 [11] Nabel K G, Clark S A, Shankar S, et al. Structural basis for continued antibody evasion by the SARS-CoV-2 receptor binding domain. Science, 2022; 375(6578): eabl6251. doi: 10.1126/science.abl6251 [12] Baptista A M, Martel P J, Petersen S B. Simulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titration. Proteins, 1997; 27(4): 523-544. doi: 10.1002/(SICI)1097-0134(199704)27:4<523::AID-PROT6>3.0.CO;2-B [13] Zhou Z, Yang Z, Ou J, et al. Temperature dependence of the SARS-CoV-2 affinity to human ACE2 determines COVID-19 progression and clinical outcome. Comput Struct Biotechnol J, 2021; 19: 161-167. doi: 10.1016/j.csbj.2020.12.005 [14] Wei G, Xi W, Nussinov R, et al. Protein ensembles: how does nature harness thermodynamic fluctuations for life? the diverse functional roles of conformational ensembles in the cell. Chem Rev, 2016; 116(11): 6516-6551. doi: 10.1021/acs.chemrev.5b00562 [15] Yao X Q, Hamelberg D. Detecting functional dynamics in proteins with comparative perturbed-ensembles analysis. Acc Chem Res, 2019; 52(12): 3455–3464 doi: 10.1021/acs.accounts.9b00485 [16] Van Wart A T, Durrant J, Votapka L, et al. Weighted implementation of suboptimal paths (WISP): an optimized algorithm and tool for dynamical network analysis. J Chem Theory Comput, 2014; 10(2): 511-517. doi: 10.1021/ct4008603 [17] Wang P, Casner R G, Nair M S, et al. Increased resistance of SARS-CoV-2 variant P. 1 to antibody neutralization. Cell Host Microbe, 2021; 29(5): 747-751. e4. doi: 10.1016/j.chom.2021.04.007 [18] Garrett M E, Galloway J G, Wolf C, et al. Comprehensive characterization of the antibody responses to SARS-CoV-2 Spike protein finds additional vaccine-induced epitopes beyond those for mild infection. Elife, 2022; 11: e73490. doi: 10.7554/eLife.73490 [19] Serapian S A, Marchetti F, Triveri A, et al. The answer lies in the energy: how simple atomistic molecular dynamics simulations may hold the key to epitope prediction on the fully glycosylated SARS-CoV-2 spike protein. J Phys Chem Lett, 2020; 11(19): 8084-8093. doi: 10.1021/acs.jpclett.0c02341 [20] Crooke S N, Ovsyannikova I G, Kennedy R B, et al. Immunoinformatic identification of B cell and T cell epitopes in the SARS-CoV-2 proteome. Sci Rep, 2020; 10(1): 14179. doi: 10.1038/s41598-020-70864-8 -

 下载:
下载:
 点击查看大图
点击查看大图
计量
- 文章访问数: 2102
- HTML全文浏览量: 1133
- PDF下载量: 35
- 被引次数: 0

