 投稿系统
投稿系统

Growth differentiation factor 11 promotes macrophage polarization towards M2 to attenuate myocardial infarction via inhibiting Notch1 signaling pathway
doi: 10.2478/fzm-2023-0008
-
Abstract:
Background Myocardial infarctions (MI) is a major threat to human health especially in people exposed to cold environment. The polarization of macrophages towards different functional phenotypes (M1 macrophages and M2 macrophages) is closely related to MI repairment. The growth differentiation factor 11 (GDF11) has been reported to play a momentous role in inflammatory associated diseases. In this study, we examined the regulatory role of GDF11 in macrophage polarization and elucidated the underlying mechanisms in MI. Methods In vivo, the mice model of MI was induced by permanent ligation of the left anterior descending coronary artery (LAD), and mice were randomly divided into the sham group, MI group, and MI+GDF11 group. The protective effect of GDF11 on myocardial infarction and its effect on macrophage polarization were verified by echocardiography, triphenyl tetrazolium chloride staining and immunofluorescence staining of heart tissue. In vitro, based on the RAW264.7 cell line, the effect of GDF11 in promoting macrophage polarization toward the M2 type by inhibiting the Notch1 Signaling pathway was validated by qRT-PCR, Western blot, and flow cytometry. Results We found that GDF11 was significantly downregulated in the cardiac tissue of MI mice. And GDF11 supplementation can improve the cardiac function. Moreover, GDF11 could reduce the proportion of M1 macrophages and increase the accumulation of M2 macrophages in the heart tissue of MI mice. Furthermore, the cardioprotective effect of GDF11 on MI mice was weakened after macrophage clearance. At the cellular level, application of GDF11 could inhibit the expression of M1 macrophage (classically activated macrophage) markers iNOS, interleukin (IL)-1β, and IL-6 in a dose-dependent manner. In contrast, GDF11 significantly increased the level of M2 macrophage markers including IL-10, CD206, arginase 1 (Arg1), and vascular endothelial growth factor (VEGF). Interestingly, GDF11 could promote M1 macrophages polarizing to M2 macrophages. At the molecular level, GDF11 significantly down-regulated the Notch1 signaling pathway, the activation of which has been demonstrated to promote M1 polarization in macrophages. Conclusions GDF11 promoted macrophage polarization towards M2 to attenuate myocardial infarction via inhibiting Notch1 signaling pathway. -
Key words:
- myocardial infarction /
- growth differentiation factor 11 /
- M1 macrophage /
- M2 macrophage /
- Notch1
-

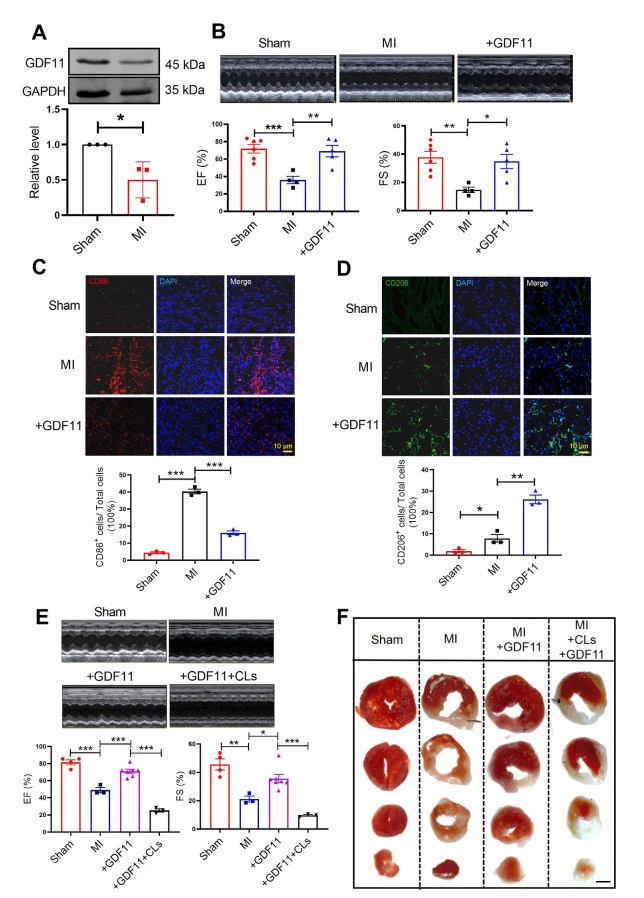
Figure 1. GDF11 regulates macrophage polarization to improve cardiac function in MI mice
(A) Western blotting detected the protein expression of GDF11 in MI, N = 3 per group. (B) Echocardiography of MI mice and GDF11-treatment mice at 7 days after MI or sham operation, N = 4-6 per group. (C) Co-immunostaining of cardiac tissue sections at 3 days after MI stained with the M1 macrophage surface marker CD86 (red) and DAPI (blue). N = 3 per group. (D) Co-immunostaining of cardiac tissue sections at 7 days after myocardial infarction stained with the M2macrophage surface marker CD206 (red) and DAPI (blue). N = 3 per group. (E) Echocardiography was measured, EF% and FS% were detected in sham mice, MI mice, MI+GDF11+CLs mice and MI+GDF11 mice, N = 3-7 per group. (F) TTC staining to verify the infarct area of myocardial infarction mice. Data are described as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; GDF11, growth differentiation factor 11; MI, myocardial infarctions; CLs, clodronate liposomes; EF, ejection fraction; FS, fractional shortening.

Figure 2. Concentration gradient studies of GDF11-induced expression of iNOS, IL-1β and IL-6 in RAW264.7 macrophages
(A-C) Cultured RAW264.7 macrophages were treated with GDF11 (1, 10, 50 ng/mL) for 24 hours. GDF11-induced the expression of several genes labeled M1 macrophages, including iNOS, IL-1β, and IL-6 were analyzed by qRT-PCR. N = 3-9 per group. (D) Cultured RAW264.7 macrophages were treated with GDF11 (50 ng/mL) for 48 hours. iNOS protein levels were measured using western blotting analysis. N = 6 per group. Data are described as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 vs. control group; GDF11, growth differentiation factor 11; IL, interleukin.<

Figure 3. Concentration gradient studies of GDF11-induced expression of Arg1, IL-10 and CD206 in RAW264.7 macrophages
(A-C) Cultured RAW264.7 macrophages were treated with GDF11 (1, 10, 50 ng/mL) for 24 hours. GDF11-induced expression of several genes labeled M2 macrophages, including Arg1, IL-10, and CD206 were analyzed by Real-time PCR. N = 3-10 per group. (D) Cultured RAW264.7 macrophages were treated with GDF11 (50 ng/mL) for 48 hours. Arg1 protein levels were measured using western blotting analysis. N = 9 per group. (E) Cultured RAW264.7 macrophages were treated with GDF11 (50 ng/mL) for 48 hours and the number of CD206+ cells was measured by flow cytometry analysis. N = 3 per group. Data are described as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 vs. control group; GDF11, growth differentiation factor 11; IL, interleukin; Arg1, arginase 1.

Figure 4. GDF11 inhibits LPS-induced inflammatory response in RAW264.7
(A-C) The mRNA levels of iNOS, IL-1β and IL-6 in the LPS (100 ng/mL) treated macrophages at the presence or absence of GDF11 (50 ng/mL). N = 3-5 per group. (D) Levels of iNOS was tested by Immunofluorescence (iNOS was shown in red, nucleus was stained by DAPI and shown in blue). (E-F) iNOS protein levels were measured using western blotting analysis. N = 4 per group. (G) The number of CD206+ cells was measured byflow cytometry analysis. N = 4 per group. Data are described as means ± SEM. **P < 0.01; ***P < 0.001; GDF11, growth differentiation factor 11; IL, interleukin.

Figure 5. GDF11 enhances anti-inflammatory reaction in LPS treated RAW264.7 cells
(A-C) The mRNA levels of Arg1, VEGF and IL-10 in the LPS treated macrophages at the presence or absence of GDF11. N = 4-6 per group. (D) The protein level of Arg1 was verified by western blotting. N = 4 per group. Data are described as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; GDF11, growth differentiation factor 11; IL, interleukin; Arg1, arginase 1; VEGF, vascular endothelial growth factor.

Figure 6. GDF11 inhibits the activation of Notch1 signaling pathway
(A)The mRNA levels of Notch1 in the LPS treated macrophages at the presence or absence of GDF11. N = 8 per group. (B-E) The mRNA levels of Notch1, Hes1, Hey1 and iNOS in the VPA treated macrophages at the presence or absence of GDF11. N = 3-7 per group. Data are described as means ± SEM (N = 3/group). *P < 0.05; **P < 0.01; ***P < 0.001; GDF11, growth differentiation factor 11; IL, interleukin; Arg1, arginase 1; VEGF, vascular endothelial growth factor; VPA, valproic acid.
-
[1] Pettersson H, Olsson D, Järvholm B. Occupational exposure to noise and cold environment and the risk of death due to myocardial infarction and stroke. Int Arch Occup Environ Health, 2020; 93(5): 571-575. doi: 10.1007/s00420-019-01513-5 [2] Claeys M J, Rajagopalan S, Nawrot T S, et al. Climate and environmental triggers of acute myocardial infarction. Eur Heart J, 2017; 38(13): 955-960. [3] Prabhu S D, Frangogiannis N G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res, 2016; 119(1): 91-112. doi: 10.1161/CIRCRESAHA.116.303577 [4] Frangogiannis N G. Regulation of the inflammatory response in cardiac repair. Circ Res, 2012; 110(1): 159-173. doi: 10.1161/CIRCRESAHA.111.243162 [5] Tsujioka H, Imanishi T, Ikejima H, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol, 2009; 54(2): 130-138. doi: 10.1016/j.jacc.2009.04.021 [6] Castro-Dopico T, Fleming A, Dennison T W, et al. Gm-Csf calibrates macrophage defense and wound healing programs during intestinal infection and inflammation. Cell Rep, 2020; 32(1): 107857. doi: 10.1016/j.celrep.2020.107857 [7] Wang Y, Smith W, Hao D, et al. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int Immunopharmacol, 2019; 70: 459-466. doi: 10.1016/j.intimp.2019.02.050 [8] Pan Y, Hui X, Hoo R L C, et al. Adipocyte-secreted exosomal microrna-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J Clin Invest, 2019; 129(2): 834-849. doi: 10.1172/JCI123069 [9] Suh J, Kim N K, Lee S H, et al. Gdf11 promotes osteogenesis as opposed to Mstn, and follistatin, a Mstn/Gdf11 inhibitor, increases muscle mass but weakens bone. Proc Natl Acad Sci U S A, 2020; 117(9): 4910-4920. doi: 10.1073/pnas.1916034117 [10] Scudellari M. Ageing research: blood to blood. Nature, 2015; 517(7535): 426-429. doi: 10.1038/517426a [11] Loffredo F S, Steinhauser M L, Jay S M, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell, 2013; 153(4): 828-839. doi: 10.1016/j.cell.2013.04.015 [12] Sinha M, Jang Y C, Oh J, et al. Restoring systemic Gdf11 levels reverses age-related dysfunction in mouse skeletal muscle. Science, 2014; 344(6184): 649-652. doi: 10.1126/science.1251152 [13] Kaiser J. Regenerative medicine. 'rejuvenating' protein doubted. Science, 2015; 348(6237): 849. doi: 10.1126/science.348.6237.849 [14] Mei W, Xiang G, Li Y, et al. Gdf11 Protects against Endothelial injury and reduces atherosclerotic lesion formation in apolipoprotein E-null mice. Mol Ther, 2016; 24(11): 1926-1938. doi: 10.1038/mt.2016.160 [15] Wang L, Wang Y, Wang Z, et al. Growth differentiation factor 11 ameliorates experimental colitis by inhibiting Nlrp3 inflammasome activation. Am J Physiol Gastrointest Liver Physiol, 2018; 315(6): G909-G920. doi: 10.1152/ajpgi.00159.2018 [16] Li W, Wang W, Liu L, et al. Gdf11 antagonizes tnf-alpha-induced inflammation and protects against the development of inflammatory arthritis in mice. FASEB J, 2019; 33(3): 3317-3329. doi: 10.1096/fj.201801375RR [17] Li Q, Jiao L, Shao Y, et al. Topical Gdf11 accelerates skin wound healing in both type 1 and 2 diabetic mouse models. Biochem Biophys Res Commun, 2020; 529(1): 7-14. doi: 10.1016/j.bbrc.2020.05.036 [18] Wang Z, Li Y, Kong D, et al. Cross-talk between mirna and notch signaling pathways in tumor development and progression. Cancer Lett, 2010; 292(2): 141-148. doi: 10.1016/j.canlet.2009.11.012 [19] Xu H, Zhu J, Smith S, et al. Notch-Rbp-J signaling regulates the transcription factor Irf8 to promote inflammatory macrophage polarization. Nat Immunol, 2012; 13(7): 642-650. doi: 10.1038/ni.2304 [20] Huang Y, Tian C, Li Q, et al. Tet1 knockdown inhibits porphyromonas gingivalis Lps/Ifn-gamma-induced M1 macrophage polarization through the Nf-Kappab pathway in Thp-1 Cells. Int J Mol Sci, 2019; 20(8): 2023. doi: 10.3390/ijms20082023 [21] Maniati E, Bossard M, Cook N, et al. Crosstalk between the canonical Nf-Kappab and notch signaling pathways inhibits ppargamma expression and promotes pancreatic cancer progression in mice. J Clin Invest, 2011; 121(12): 4685-4699. doi: 10.1172/JCI45797 [22] Zhang H, Hilton M J, Anolik J H, et al. Notch inhibits osteoblast formation in inflammatory arthritis via noncanonical Nf-Kappab. J Clin Invest, 2014; 124(7): 3200-3214. doi: 10.1172/JCI68901 [23] Sun W, Zhang H, Wang H, et al. Targeting notch-activated m1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J Bone Miner Res, 2017; 32(7): 1469-1480. doi: 10.1002/jbmr.3117 [24] Li Q, He X, Yu Q, et al. The notch signal mediates macrophage polarization by regulating Mir-125a/Mir-99b expression. Artif Cells Nanomed Biotechnol, 2019; 47(1): 833-843. doi: 10.1080/21691401.2019.1576711 [25] Leuschner F, Rauch P J, Ueno T, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med, 2012; 209(1): 123-137. doi: 10.1084/jem.20111009 [26] Yang J, Kong C, Jia L, et al. Association of accelerated long-term forgetting and senescence-related blood-borne factors in asymptomatic individuals from families with autosomal dominant alzheimer's disease. Alzheimers Res Ther, 2021; 13(1): 107. doi: 10.1186/s13195-021-00845-0 [27] Dou F, Wu B, Chen J, et al. Pparalpha targeting Gdf11 inhibits vascular endothelial cell senescence in an atherosclerosis model. Oxid Med Cell Longev, 2021; 2021: 2045259. [28] Lehallier B, Shokhirev M N, Wyss-Coray T, et al. Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell, 2020; 19(11): e13256. [29] Li L, Wei X, Wang D, et al. Positive effects of a young systemic environment and high growth differentiation factor 11 levels on chondrocyte proliferation and cartilage matrix synthesis in old mice. Arthritis Rheumatol, 2020; 72(7): 1123-1133. doi: 10.1002/art.41230 [30] Egerman M A, Glass D J. The role of Gdf11 in aging and skeletal muscle, cardiac and bone homeostasis. Crit Rev Biochem Mol Biol, 2019; 54(2): 174-183. doi: 10.1080/10409238.2019.1610722 [31] Stengel S, Quickert S, Lutz P, et al. Peritoneal level of Cd206 associates with mortality and an inflammatory macrophage phenotype in patients with decompensated cirrhosis and spontaneous bacterial peritonitis. Gastroenterology, 2020; 158(6): 1745-1761. doi: 10.1053/j.gastro.2020.01.029 [32] Gharib S A, McMahan R S, Eddy W E, et al. Transcriptional and functional diversity of human macrophage repolarization. J Allergy Clin Immunol, 2019; 143(4): 1536-1548. doi: 10.1016/j.jaci.2018.10.046 [33] Sajti E, Link V M, Ouyang Z, et al. Transcriptomic and epigenetic mechanisms underlying myeloid diversity in the lung. Nat Immunol, 2020; 21(2): 221-231. doi: 10.1038/s41590-019-0582-z [34] Pan H, Huang W, Wang Z, et al. The Ace2-Ang-(17)-Mas axis modulates M1/M2 macrophage polarization to relieve Clp-induced inflammation via Tlr4-mediated Nf-small Ka, cyrillicb and mapk pathways. J Inflamm Res, 2021; 14: 2045-2060. doi: 10.2147/JIR.S307801 [35] Li Y, Dong M, Wang Q, et al. Himf deletion ameliorates acute myocardial ischemic injury by promoting macrophage transformation to reparative subtype. Basic Res Cardiol, 2021; 116(1): 30. doi: 10.1007/s00395-021-00867-7 [36] Jiao L, Shao Y, Yu Q, et al. Gdf11 replenishment protects against hypoxia-mediated apoptosis in cardiomyocytes by regulating autophagy. Eur J Pharmacol, 2020; 885: 173495. doi: 10.1016/j.ejphar.2020.173495 -

 下载:
下载:






 点击查看大图
点击查看大图
计量
- 文章访问数: 686
- HTML全文浏览量: 425
- PDF下载量: 46
- 被引次数: 0

