 投稿系统
投稿系统

Overexpression of microRNA-135b-5p attenuates acute myocardial infarction injury through its antioxidant and anti-apptotic properties
doi: 10.2478/fzm-2021-0011
-
Abstract:
Objective: Myocardial infarction (MI) remains the leading cause of morbidity and mortality due partly to the limited regenerative capacity of cardiomyocytes to replace cardiomyocyte lost due to apoptosis. Inhibiting cardiomyocyte apoptosis is recognized as an effective therapeutic approach for MI. MicroRNAs (miRNAs, miRs), which regulate target genes at the post-transcriptional level, play a significant role in the regulation of cardiovascular diseases such as MI. MicroRNA-135b (miR-135b) has a protective effect on cardiomyocytes. However, the role of miR- 135b in cardiomyocyte apoptosis in infarct myocardium needs further clarification. Methods: We generated α-MHC-miR-135b transgenic mice to investigate the role of miR-135b in myocardial injury after MI. MiR- 135b mimic and negative control (NC) were transfected into H2O2-induced cardiomyocytes to evaluate the effect of overexpression of miR-135b on the levels of reactive oxygen species (ROS) and apoptosis. Results: Our results showed that overexpression of miR-135b had protective effect on cardiomyocyte injury both in vivo and in vitro. MiR-135b inhibited cardiomyocyte apoptosis and ROS generation, downregulated proapoptosis proteins (cleaved-caspase-3 and Bax), and increased antiapoptosis protein (Bcl-2). Moreover, miR-135b showed an inhibitory effect on apoptosis-related protein target transient receptor potential vanilloid-type 4 (TRPV4) cation channel. Conclusion: MiR-135b might be considered a new molecular target for potential replacement therapy as antiapoptotic cardioprotection in the setting of MI.
-
Key words:
- myocardial infarction /
- apoptosis /
- microRNA-135b /
- TRPV4
-

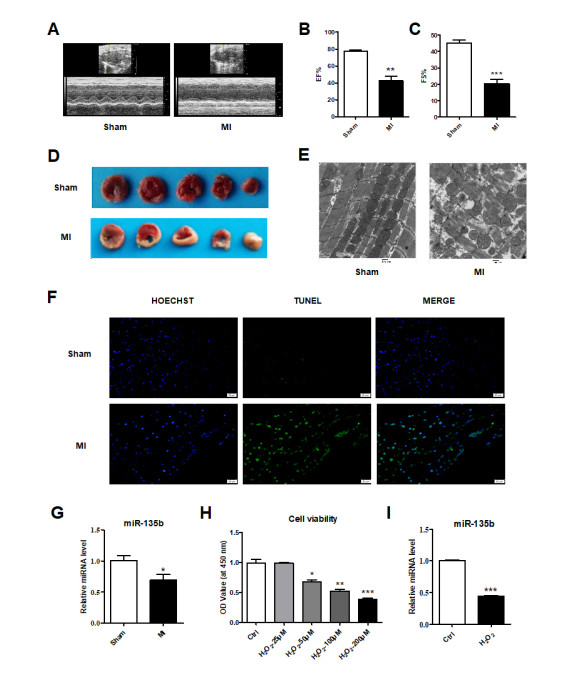
Figure 1. MI induced functional and morphological changes in mouse hearts
(A) M-mode echocardiograms. (B) Ejection fraction and (C) Fractional shortening. (D) TTC staining results. (E) Election microscope images. Scale bars = 500 nm. (F) TUNEL staining results. The mRNA expression of miR-135b (G) in vivo and (I) in vitro. (H) CCK8 assay result. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham or Ctrl. The data are presented as mean ± SD.

Figure 2. Overexpression of miR-135b in transgenic (Tg) mice reverses MI-induced impairments of cardiac function
(A) The mRNA expression of miR-135b in vitro. (B) M-mode echocardiograms. (C) Ejection fraction and (D) Fractional shortening. (E) TTC staining results. (F) Election microscope images. Scale bars = 500 nm. (G) TUNEL staining results. n = 3; ***P < 0.001 vs. WT, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. WT+MI. &&&P < 0.001 vs. Tg+MI. The data are presented as mean ± SD.

Figure 3. Forced expression of miR-135b suppresses ROS generation and apoptosis in primary cultured neonatal mouse ventricular cells (NMVCs)
(A) The mRNA expression of miR-135b in vitro. (B) CCK8 assay result. (C) Flow cytometry results and (D) corresponding statistical result in vitro. (E) ROS generation in vitro. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. Ctrl or NC, ###P < 0.001 vs. H2O2. The data are presented as mean ± SD.

Figure 4. Overexpression of miR-135b alters the expression of apoptosis-related protein regulators in Tg mice
The mRNA expression of (A) Caspase-3 (B) Bax (C) Bcl-2 and (D) Bax/Bcl-2 mRNA ratio in vitro. The protein expression of (E) Caspase-3 (F) Bax (G) Bcl-2 and (H) Bax/Bcl-2 protein ratio in vitro. n = 3; **P < 0.01, ***P < 0.001 vs. WT, ##P < 0.01, ###P < 0.001 vs. WT+MI. The data are presented as mean ± SD.

Figure 5. Forced expression of miR-135b alters the expression of apoptosis-related protein regulators in cultured NMVCs
The mRNA expression of (A) Caspase-3 (B) Bax (C) Bcl-2 and (D) Bax/Bcl-2 mRNA ratio in vitro. The protein expression of (E) Caspase-3 (F) Bax (G) Bcl-2 and (H) Bax/Bcl-2 protein ratio in vitro. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. WT+MI. The data are presented as mean ± SD.

Figure 6. miR-135b represses the expression of TRPV4 both in vivo and in vitro
(A) The predicted binding site for miR-135b in TRPV4 sequences of humans and mice. (B) The mRNA and (C) protein expression of TRPV4 in vivo. (D) The mRNA and (E) protein expression of TRPV4 in vitro. n = 3; **P < 0.01 vs. WT or Ctrl, #P < 0.05, ##P < 0.01 vs. WT+MI or H2O2. The data are presented as mean ± SD.
-
[1] Friedman P L, Fenoglio J J, Wit A L. Time course for reversal of electrophysiological and ultrastructural abnormalities in subendocardial Purkinje fibers surviving extensive myocardial infarction in dogs. Circ Res, 1975; 36(1): 127-144. doi: 10.1161/01.RES.36.1.127 [2] Spear J F, Michelson E L, Spielman S R, et al. The origin of ventricular arrhythmias 24 hours following experimental anterior septal coronary artery occlusion. Circulation, 1977; 55(6): 844-852. doi: 10.1161/01.CIR.55.6.844 [3] Frangogiannis N G. Pathophysiology of myocardial infarction. Compr Physiol, 2015; 5(4): 1841-1875. [4] Zhang J, Qiu W, Ma J, et al. miR-27a-5p attenuates hypoxia-induced rat cardiomyocyte injury by inhibiting Atg7. Int J Mol Sci, 2019; 20(10): 2418. doi: 10.3390/ijms20102418 [5] Sun T, Dong Y H, Du W, et al. The role of microRNAs in myocardial infarction: From molecular mechanism to clinical application. Int J Mol Sci, 2017; 18(4): 745. doi: 10.3390/ijms18040745 [6] Li A, Yu Y, Ding X, et al. MiR-135b protects cardiomyocytes from infarction through restraining the NLRP3/Caspase-1/IL-1β pathway. Int J Cardiol, 2020; 307(2): 137-145. http://www.sciencedirect.com/science/article/pii/S016752731932176X [7] Chen C, Shen H, Huang Q, et al. The circular RNA CDR1as regulates References the proliferation and apoptosis of human cardiomyocytes through the miR- 135a/HMOX1 and miR-135b/HMOX1 axes. Genet Test Mol Biomarkers, 2020; 24(9): 537-548. doi: 10.1089/gtmb.2020.0034 [8] Grace M S, Bonvini S J, Belvisi M G, et al. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol Ther, 2017; 177: 9-22. doi: 10.1016/j.pharmthera.2017.02.019 [9] Che H, Xiao G S, Sun H Y, et al. Functional Trpv2 and Trpv4 channels in human cardiac C-Kit progenitor cells. J Cell Mol Med, 2016; 20(6): 1118- 1127. doi: 10.1111/jcmm.12800 [10] Qi, Y. Li Z, Kong C, et al. Uniaxial cyclic stretch stimulates Trpv4 to induce realignment of human embryonic stem cell-derived cardiomyocytes. J Mol Cell Cardiol, 2015; 87: 65-73. doi: 10.1016/j.yjmcc.2015.08.005 [11] Zhao, Y, Huang H, Jiang Y, et al. Unusual localization and translocation of Trpv4 protein in cultured ventricular myocytes of the neonatal rat. Eur J Histochem, 2012; 56(3): e32. doi: 10.4081/ejh.2012.e32 [12] Dong Q, Li J, Wu Q F, et al. Blockage of transient receptor potential vanilloid 4 alleviates myocardial ischemia/reperfusion injury in mice. Sci Rep, 2017; 7(1): 42678. doi: 10.1038/srep42678 [13] Wu Q F, Qian C, Zhao N, et al. Activation of transient receptor potential vanilloid 4 involves in hypoxia/reoxygenation injury in cardiomyocytes. Cell Death Dis, 2017; 8(5): e2828. doi: 10.1038/cddis.2017.227 [14] Gao E, Koch W J. A novel and efficient model of coronary artery ligation in the mouse, Methods Mol. Biol, 2013; 1037: 299-311. http://www.ncbi.nlm.nih.gov/pubmed/24029943 [15] He H, Liu X, Lv L, et al. Calcineurin suppresses AMPKdependent cytoprotective autophagy in cardiomyocytes under oxidative stress, Cell Death Dis, 2014, 5(1): e997. doi: 10.1038/cddis.2013.533 [16] Lu Q, Zemskov E A, Sun X, et al. Activation of the mechanosensitive Ca2+ channel TRPV4 induces endothelial barrier permeability via the disruption of mitochondrial bioenergetics. Redox Biol, 2021; 38(6): 101785. [17] Konstantinidis K, Whelan R S, Kitsis R N. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol, 2012; 32(7): 1552-1562. doi: 10.1161/ATVBAHA.111.224915 [18] Kajstura J, Cheng W, Reiss K, et al. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest, 1996; 74(1): 86-107. [19] Xie X J, Fan D M, Xi K, et al. Suppression of microRNA-135b-5p protects against myocardial ischemia/reperfusion injury by activating JAK2/ STAT3 signaling pathway in mice during sevoflurane anesthesia. Biosci Rep, 2017; 37(3): BSR20170186. doi: 10.1042/BSR20170186 [20] Zhu H J, Wang D G, Yan J, et al. Up-regulation of microRNA-135a protects against myocardial ischemia/reperfusion injury by decreasing TXNIP expression in diabetic mice. Am J Transl Res, 2015; 7(12): 2661- 2671. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4731664/pdf/ajtr0007-2661.pdf [21] Adapala R K, Kanugula A K, Paruchuri S, et al. TRPV4 deletion protects heart from myocardial infarction induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Res Cardiol, 2020; 115(2): 14. doi: 10.1007/s00395-020-0775-5 [22] Chaigne S, Cardouat G, Louradour J, et al. Transient receptor potential vanilloid 4 channel participates in mouse ventricular electrical activity. Am J Physiol Heart Circ Physiol, 2021; 320(3), doi: 10.1152/ajpheart.00497.2020. [23] Adapala R K, Thoppil R J, Luther D J, et al. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol, 2013; 54: 45-52. doi: 10.1016/j.yjmcc.2012.10.016 [24] Everaerts W, Zhen X, Ghosh D, et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci USA, 2010; 107(44): 19084-19089. doi: 10.1073/pnas.1005333107 [25] Thorneloe K S, Cheung M, Bao W, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med, 2012; 4(159) http://www.onacademic.com/detail/journal_1000038014919010_98ee.html -

 下载:
下载:





 点击查看大图
点击查看大图
计量
- 文章访问数: 793
- HTML全文浏览量: 320
- PDF下载量: 14
- 被引次数: 0

